
Neuronal longevity
Mature neurons normally live throughout the life of an organism (e.g., years), as their continuous survival is vital for learning and memory, and mammalian brains have limited regeneration capability. Apparently necessary (though insufficient) for the lifelong survival of neurons is sustained suppression of apoptosis, a ubiquitous regulated cell death pathway controlling cell turnover and tissue homeostasis in metazoans. Non-neural cells and tissues readily engage apoptosis throughout life in response to intrinsic and extrinsic stress. This cellular suicidal program needs to be reined in for post-mitotic neurons. While neuronal apoptosis has been studied for its activation in diseases and after lethal insults, the genetic regulation enforcing restriction of neuronal apoptosis is largely unknown. a robust genetic program that intrinsically enables neuronal longevity.
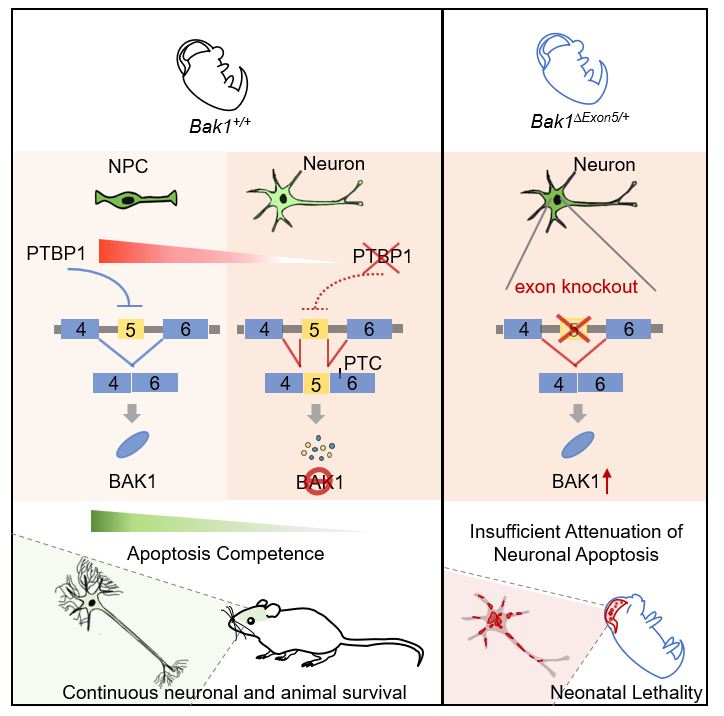
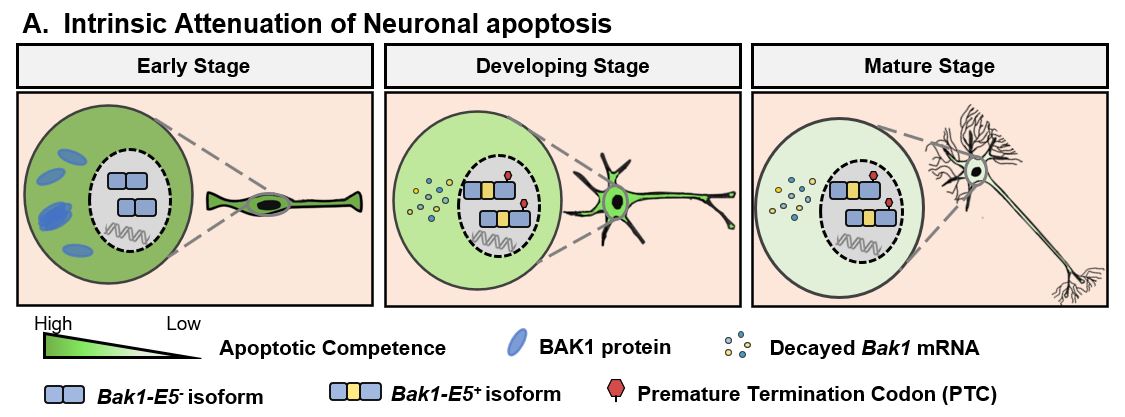
We discovered a robust genetic program that intrinsically attenuate apoptosis to enable neuronal longevity. Developmental downregulation of the splicing regulator PTBP1 in immature neurons allows neural-specific splicing of the evolutionarily-conserved Bak1 exon 5. Exon 5 inclusion triggers nonsense-mediated mRNA decay and unproductive translation of Bak1 transcripts, leading to suppression of pro-apoptotic BAK1 proteins and allowing neurons to reduce apoptosis. Germline heterozygous ablation of exon 5 increases BAK1 proteins exclusively in the brain, inflates neuronal apoptosis, and leads to early postnatal mortality. Therefore, neural-specific exon 5 splicing and depletion of BAK1 proteins are necessary for uninterrupted neuronal survival. While apoptosis is important for development, attenuation of apoptosis competence through neural-specific alternative splicing of Bak1 is essential for neuronal longevity and animal survival.
Previous studies have highlighted extrinsic signals in supporting neuronal survival. Two of the best studied extrinsic survival signals are neurotrophins in the peripheral nervous system and synaptic activity in the central nervous system. The “neurotrophic theory” and the “activity-dependent survival theory” assume the default cell fate of neurons is death and emphasize that neuronal survival is directed by target cells/tissues. Specifically, to survive, neurons need target-derived trophic factors in the peripheral nervous system and/or appropriate synaptic transmission in the central nervous system.
Our findings complement these theories and show neuronal survival is also intrinsically controlled by apoptosis attenuation. We show apoptosis sensitivity of cortical neurons progressively decreases during differentiation even before synapse formation. Apoptotic resistance therefore is not a “toggle switch” imposed only by fruitful neuronal connections (or reaching target tissues/cells). Since a post-mitotic neuron’s lifespan includes the time spent as an immature neuron and even immature neurons need to live weeks (in rodents) and months (in humans) before forming functional neural circuits, programming of apoptosis resistance apparently starts as early as neuron birth. loss of BAK1 is like losing the “fuel” of apoptosis (i.e., apoptosis attenuation). Turning off apoptosis effectors appears to be an effective strategy to proactively evade apoptosis, and we show eliminating BAK1 in neurons is essential for this purpose.
The genetic necessity of apoptosis attenuation for animal survival is clearly demonstrated by Bak1∆exon5/+ mice. Exon 5 is spliced exclusively in neural tissues leading to neural-specific “gene knockout” of BAK1 to reduce neuronal apoptosis. This regulation is unlikely the sole mechanism supporting continuous neuronal survival but is so essential that heterozygous deletion of its central element (Bak1 exon 5) is enough to increase neuronal apoptosis and cause Bak1∆exon5/+ animal lethality.
Comparison between Bak1∆exon5/+ and Bak1−/− mice is interesting. Bak1−/− mice with homozygous deletion of exons 2-6 are viable, fertile, and have no detectable phenotypic abnormalities (Lindsten et al. 2000). In contrast, Bak1∆exon5/+ mice (with the deletion of exon 5 in a single allele) cannot survive. The severer phenotype in Bak1∆exon5/+ mice is not due to the size of deletion but demonstrates the intricate regulation of genetic elements through the control of RNA processing. As exon 5 does not encode the regular BAK1 protein, it showcases essential roles of regulatory elements outside of protein coding sequences. Our findings also solidify the physiological significance of AS-NMD regulatory mechanism (Zheng, 2016).
The phenotypic contrast originates from the opposite impacts of their genetic deletions on BAK1 protein expression. Bak1−/− eliminates BAK1 protein but has no phenotypes because of the negligible BAK1 expression in the brain and BAX compensation in peripheral tissues. Bak1∆exon5/+ does not affect peripheral tissues but substantially increases BAK1 protein expression in the brain. The latter leads to increased neuronal apoptosis and early mortality. A small percentage of triple knockout mice of Bak1 and its close paralogs Bax and Bok appeared largely normal, suggesting mouse development or survival do not absolutely require activation of apoptosis. The phenotypes of Bak1∆exon5/+ mice, on the other hand, show that apoptosis suppression in neurons is necessary for mouse survival.